Comparing a pair of genomes, say, to identify a new sample as a pathogen or not, can be done fairly quickly. But if you need pairwise comparisons among a large group of genomes, like for building trees of their ancestry, the problem becomes quadratic and quickly explodes.
Inspired by an algorithm for duplicate detection in web search, we used locality sensitive hashing to preprocess thousands of genomes, creating small signatures of each called sketches. Comparing these sketches is orders of magnitude faster than the whole genomes, making the quadratic step of pairwise comparison much more tractable. This allowed us to perform an unprecedented clustering, shown below, of every organism in RefSeq, the major repository of reference genome sequences.

The sketching strategy also vastly reduces the memory footprint of searching a single sample against a large database, such as RefSeq, allowing real time search on limited hardware, as described in this talk:
Containment
What if, instead of comparing two similarly-sized genomes, we want to ask whether a small sequence (like a microbial genome) is contained in a much larger sequence (e.g. a metagenome)? It turns out the original algorithm doesn’t work so well for this case. This motivated us to adapt it into a new method that acts as a “screen.” Instead of creating a sketch for the larger sequence mixture, we stream through all of it while keeping track of hits to a database of reference sketches.
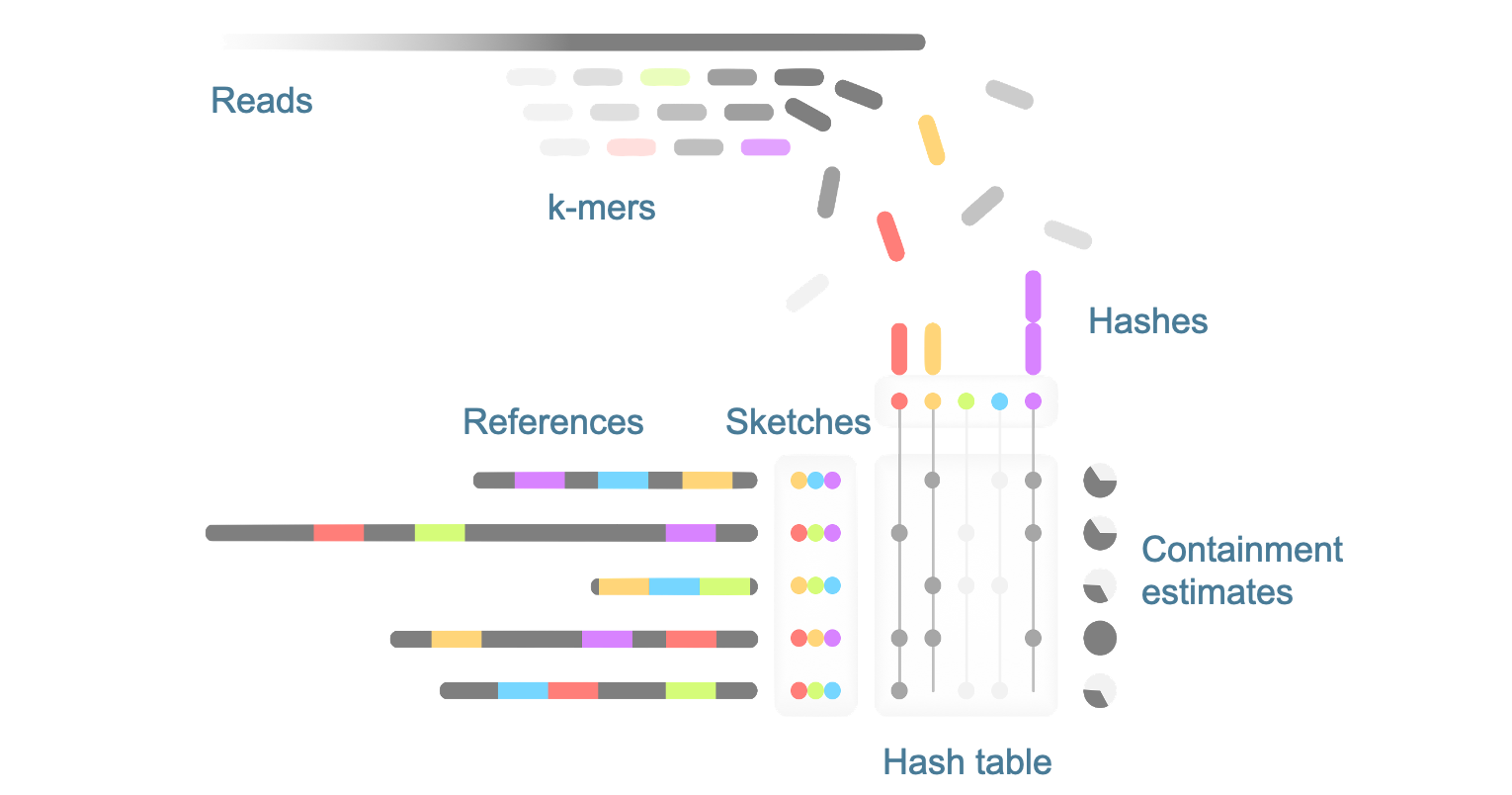
Combined with a large compute cluster, the efficiency of this approach allowed us to create an unprecedented table of the containment of all RefSeq genomes (~100,000) within every metagenome (~100,000) in the SRA (the major repository for raw sequence data). This provides a valuable resource for retrospective discovery, for example finding relatives of a newly discovered virus.
Mash: fast genome and metagenome distance estimation using MinHash
Brian Ondov, Todd Treangen, Páll Melsted, Adam Mallonee, Nicholas Bergman, Sergey Koren, Adam Phillippy
pdf • doi